Ендотеліїт у патогенезі хронічної ІХС: клінічні прояви та підходи до лікування

Резюме. Незважаючи на те що протягом останніх років у клінічній практиці активно впроваджувалися стратегії оптимізації профілактики та лікування ішемічної хвороби серця (ІХС), наслідки цього стану все ще становлять значний тягар для здоров’я людини, утримуючи провідні позиції у структурі смертності та захворюваності.
Етіологія ішемії міокарда наразі всебічно вивчається. Клінічні, ангіографічні й автопсійні дані свідчать про складну патофізіологію ІХС, що виходить за рамки загальноприйнятої та спрощеної ролі атеросклерозу. Зокрема, існує гіпотеза, що запалення, впливаючи на ендотеліальні клітини (ЕК), стимулює розвиток та еволюцію атеросклерозу, а також спричиняє виникнення гострих тромботичних ускладнень. Водночас запалення може бути не лише причиною, а й наслідком ІХС. У такому разі воно спочатку охоплює міокард і лише потім стає системним. Беззаперечно важливу роль у регуляції запальних реакцій відіграє ендотелій.
Тривалий вплив тригерних патофізіологічних стимулів на клітини ендотелію є пусковим механізмом його прозапальної активації з розвитком ендотеліїту, а надалі – ендотеліальної дисфункції, що зумовлює хронічне порушення регуляції мікроциркуляції органів і тканин із наступним розвитком ішемічних ускладнень.
В статті розкривається роль ендотеліїту та ендотеліальної дисфункції в патогенезі ІХС, демонструються їхні клінічні наслідки, а також наводяться можливості посилення патогенетичного лікування.
Ключові слова: ішемічна хвороба серця, стенокардія, ендотеліїт, ендотеліальна дисфункція, L-аргінін, L-карнітин.
Стаття. Незважаючи на те що протягом останніх років у клінічній практиці активно впроваджувалися стратегії оптимізації профілактики та лікування ішемічної хвороби серця (ІХС), наслідки цього стану все ще становлять значний тягар для здоров’я людини, утримуючи провідні позиції у структурі смертності та захворюваності. Етіологія ішемії міокарда наразі всебічно вивчається. Клінічні, ангіографічні й автопсійні дані свідчать про складну патофізіологію ІХС, що виходить за рамки загальноприйнятої та спрощеної ролі атеросклерозу. Зокрема, існує гіпотеза, що запалення, впливаючи на ендотеліальні клітини (ЕК), стимулює розвиток та еволюцію атеросклерозу, а також спричиняє виникнення гострих тромботичних ускладнень. Водночас запалення може бути не лише причиною, а й наслідком ІХС. У такому разі воно спочатку охоплює міокард і лише потім стає системним. Беззаперечно важливу роль у регуляції запальних реакцій відіграє ендотелій. Тривалий вплив тригерних патофізіологічних стимулів на клітини ендотелію є пусковим механізмом його прозапальної активації з розвитком ендотеліїту, а надалі – ендотеліальної дисфункції, що зумовлює хронічне порушення регуляції мікроциркуляції органів і тканин із наступним розвитком ішемічних ускладнень.
Ендотеліїт як причина та наслідок хронічної ІХС
Ішемічна хвороба серця (ІХС) – це патологічний процес, що в більшості випадків характеризується розвитком атеросклеротичних бляшок в епікардіальних артеріях, а в його основі лежить порушення кровотоку у судинах серця, що призводить до недостатнього кровопостачання серцевого м’яза. ІХС є хронічним, та, як правило, прогресуючим захворюванням навіть під час стабільного клінічного перебігу [1]. Беззаперечно важливу роль у розвитку та клінічних проявах атеросклеротичного ураження судин серця, як одного із провідних етіологічних чинників ІХС, відіграють запалення ендотелію (ендотеліїт) та ендотеліальна дисфункція. Відомо, що специфічне розпізнавання антигенів рецепторами імунної системи ініціює місцеву запальну відповідь проти патогенів або тканин самого господаря. Запальні реакції зазвичай є адаптивними та знищують збудника чи сприяють гомеостазу, стимулюючи видалення пошкодженої тканини й мертвих клітин фагоцитами. І навпаки, надмірна за інтенсивністю або тривалістю запальна відповідь може бути патогенною, та викликати посилення місцевого пошкодження тканини: саме такий дезадаптивний запальний процес зумовлює розвиток ендотеліїту та дисфункції ендотелію, спричиняючи цим самим розлади ендотеліальної регуляції судинного тонусу, а також може бути рушійним чинником в ініціації та прогресуванні атеросклерозу, та, як наслідок – ІХС [2, 3].
Ендотеліальна дисфункція – ключовий момент у патогенезі атеросклерозу, тоді як атеросклероз – клінічний прояв ендотеліальної дисфункції [4-6]. Як відомо, зміни в ендотеліальному моношарі відбуваються ще на ранніх стадіях атерогенезу [7]. Проте, ендотеліїт та ендотеліальна дисфункція не лише беруть участь у розвитку ІХС, передуючи атерогенезу, а й відіграють важливу роль у подальшому прогресуванні атеросклерозу та ІХС, про що зокрема свідчить підвищення рівня маркерів системного запалення, таких як С-реактивний протеїн, фактор некрозу пухлин альфа (ФНП-а), інтерлейкін-1β, інтерлейкін-6 та інтерлейкін-8 у системному кровообігу пацієнтів з ІХС, а також доведений зв’язок підвищеного рівня цих маркерів із ступенем ураження коронарного русла та дестабілізацією перебігу захворювання[8,9]. У досліджені, проведеному P. Severino, та співав. було встановлено, що локальне запалення впливає на продукцію деяких протеолітичні ферментів, що сприяє розриву атеросклеротичної бляшки [10]. Окрім цього, ендотеліїт та ендотеліальна дисфункція вважаються незалежними предикторами розвитку рестенозів після інтервенційних втручань у пацієнтів з ІХС, що було продемонстровано у дослідженні Wang і співавт [11,12].
Фактори ризику розвитку ендотеліїту
Одними з основних антигенів імунної відповіді при розвитку атеросклерозу нині вважаються окислені ліпопротеїни низької щільності (ЛПНЩ)[13,14], які відіграють фундаментальну роль у всьому процесі атерогенезу, починаючи від утворення бляшок і закінчуючи їх дестабілізацією. Окислені ЛПНЩ підвищують експресію аргінази та знижують рівень ендотеліальної NO-синтази (eNOS) в ендотеліоцитах, тим самим сприяючи зменшенню продукції оксиду азоту (NO), стимулюють утворення активних форм кисню (АФК) і посилюють адгезію моноцитів до активованих ЕК, чим зумовлюють розвиток ендотеліїту та ендотеліальної дисфункції [15,16]. Крім того, окислені ЛПНЩ можуть індукувати міграцію та проліферацію гладеньком’язових клітин судин (VSMC), активують сигнальні шляхи SR/TLR (Toll-подібний рецептор) у макрофагах, що призводить до індукції прозапальних цитокінів, зокрема інтерлейкіну-1β шляхом активації цитозольного протеїну Nod-подібного рецептора (NLRP3), а також до продукції АФК. Окислені ЛПНЩ можуть індукувати апоптоз і некроз судинних ЕК, VSMC і макрофагів [17-20]. Отже, підвищені рівні окислених ЛПНЩ пов’язані з нестабільністю бляшок і позитивно корелюють із тяжкістю ішемії міокарда при атеросклеротичних ураженнях вінцевих артерій [21]. Ці процеси модулюються надмірною експресією лектиноподібних окислених ЛПНЩ-рецепторів-1 (LOX-1) ЕК. ЕК посилюють експресію LOX-1 у відповідь на стимуляцію окислених ЛПНЩ прозапальними цитокінами та проатерогенними факторами, як-от ангіотензин II. На кінцевих етапах атерогенезу окислені ЛПНЩ спричиняють розвиток апоптотичної смерті ЕК, можливо, саме через надмірну експресію LOX-1.
LOX-1 важливі для ендотеліально-опосередкованого судинного гомеостазу та запобігання зсіданню крові у фізіологічних умовах [12]. Модуляція LOX-1 окисленими ЛПНЩ призводить до надлишкового вироблення супероксид-аніонів. У присутності NO вони зумовлюють утворенню пероксинітриту (ONOO–) – високореактивного токсичного окислювача ЕК, який спричиняє апоптоз клітин [22].
До інших чинників ризику, причинно пов’язаних з атеросклерозом, та, відповідно, із розвитком ІХС, належать артеріальна гіпертензія, куріння, гіперглікемія, ожиріння та метаболічний синдром. Хоча механізми, які пов’язують ці чинники ризику з атерогенезом, наразі залишаються не до кінця з’ясованими, всі вони також впливають на розвиток дисбалансу оксидантно-протиоксидантної системи та беруть участь в активації запальних шляхів, зумовлюючи розвиток ендотеліїту. Як наслідок – вплив атерогенних факторів ризику, в тому числі розглянутих вище, перешкоджає виробленню ЕК ендогенних вазодилататорів, зокрема NO [23], що сприяє розвитку ендотеліальної дисфункції.
Механізми розвитку ендотеліїту
Ендотелій як повноцінний орган відіграє беззаперечно важливу роль у регуляції запальних реакцій. Він не тільки служить фізичним бар’єром, який обмежує потік речовин і рідини в тканини та з них, але також виступає ключовим гравцем у регуляції гомеостазу судинного тонусу шляхом вивільнення вазодилататорних факторів, як-от NO та простациклін, і вазоконстрикторних факторів, як-от ендотелін-1 і тромбоксан А2 [24-26]. Ендотелій є джерелом широкого спектра факторів, які локально регулюють проникність, ріст і міграцію клітин, функцію тромбоцитів і запалення [24, 27]. Оскільки запалення вирішальною мірою включає адгезію, просочування судин й інфільтрацію імунних клітин, а також активацію цих інфільтрованих імунних клітин [28], стає зрозуміло, що активація ЕК може спричиняти запалення судин і, зрештою, тканин.
У фізіологічних умовах завдяки виробленню NO та простацикліну (PGI2) лейкоцити не можуть тривало прилипати до ендотелію. Під дією запалення значно змінюється взаємодія між ендотелієм і лейкоцитами, що призводить до експресії ЕК молекул адгезії, які зв’язуються з лейкоцитами, зберігаючи та підсилюючи місцеву запальну реакцію[10].
Відомо, що ендотелій реагує на запалення підвищенням проникності, розширенням судин, наростанням діапедезу лейкоцитів, коагуляції та тромбоутворення. Механізм стимуляції ендотелію залежить від тригерного агента та зазвичай пов’язаний із викидом низки медіаторів запалення й мобілізації вмісту гранул ЕК на клітинній поверхні [29,30]. Коли є потреба більш стійкої й ефективної регуляції запального процесу, реалізується наступна стадія ендотеліальної відповіді, основним механізмом якої є цитокінозалежна активація плейотропних факторів транскрипції, як-от ядерний фактор κB (NF-κB), що впливає на експресію генів і синтез білків [31]. Стимуляція ендотелію, що відбувається під дією ушкоджувальних факторів і медіаторів запалення, опосередковується рецепторами, пов’язаними з G-білком (GPCR), є тимчасовою та не призводить до стійких морфологічних і функціональних змін.
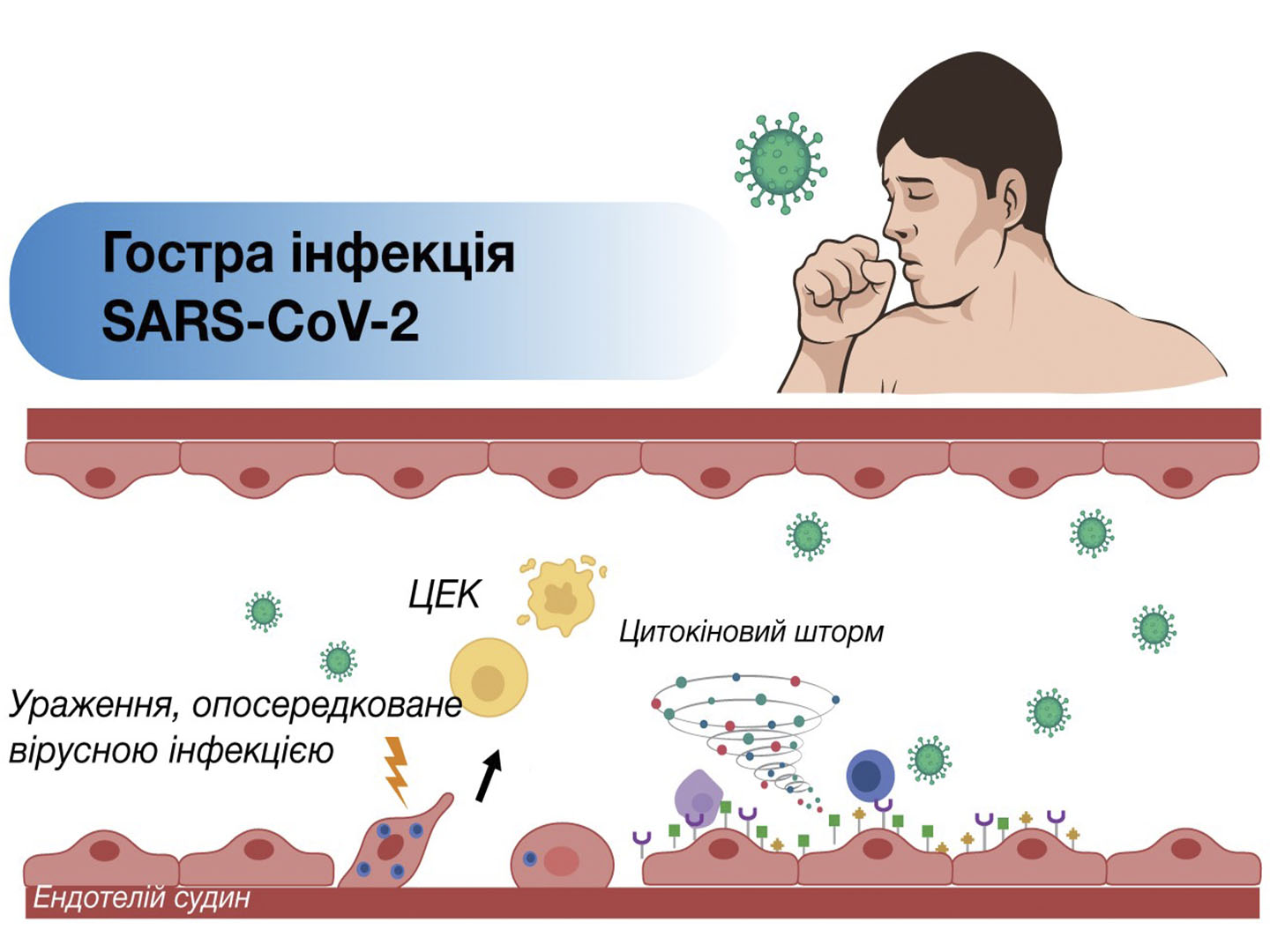
Проте, ЕК мають певний фізіологічний ліміт адаптивної реакції на дію стимулів різного ґенезу, перевищення якого неодмінно спричинятиме пошкодження ендотелію та розвиток його запалення – ендотеліїту[32]. Розвиток ендотеліїту є наслідком як прямої інвазії інфекційних патогенів, так й імунної відповіді хазяїна [33]. Термін «ендотеліїт» спочатку використовувався для опису патологічного процесу рогівки, який класично проявляється її набряком і наявністю запальних клітинних відкладень або преципітатів. Проте, пізніше було встановлено, що ендотеліїт також може розвиватися внаслідок оксидативного стресу, впливу цитокінів, ішемії/гіпоксії тканин, дисліпопротеїнемії, ендогенної та екзогенної інтоксикації, вікових змін та інших патологічних процесів, що виникають на тлі артеріальної гіпертензії, дії полютантів сигаретного диму, старіння, а також інфекцій, в тому числі вірусних [34-36]. Пряме ураження ендотеліоцитів вірусами або їх опосередковане пошкодження імунними клітинами, цитокінами та вільними радикалами може призвести до вираженої дисфункції ендотелію, що своєю чергою спричиняє порушення мікроциркуляції та вазоконстрикції з подальшим розвитком ішемії органів, запалення, набряку тканин і гіперкоагуляції [37].
Зокрема, у низці досліджень продемонстровано, що судинний ендотеліїт зумовлює системне запалення та коагулопатію, які є ключовими ланками патогенезу гострого респіраторного дистрес-синдрому, поліорганної недостатності та синдрому дисемінованого внутрішньосудинного зсідання при коронавірусній хворобі (COVID-19) [38,39]. Окрім цього, за даними J. Seeßle та співавт. (2021), через 12 міс після COVID-19 до нормального рівня здоров’я повністю повернулися тільки 22,9% пацієнтів [40]. Було встановлено, що біохімічні та запальні шляхи в організмі можуть залишатися порушеними протягом тривалого часу після стихання інфекції SARS-CoV-2 навіть у безсимптомних і помірно уражених пацієнтів [41]. Вважається, що ендотеліїт, який зберігається після гострої фази, стає однією з головних причин клінічних симптомів Long COVID [42].
Зміни ЕК реєстрували й у разі їх інфікування іншими вірусними агентами, зокрема вірусами простого герпесу 1 та 2 типів, вірусом Епштейна – Барр, цитомегаловірусом, вірусом герпесу 6 типу та вірусом грипу типу А [43-45]. Такі зміни ЕК характеризуються зниженням активності натрієвої помпи, пошкодженням щільних міжклітинних контактів, активацією вироблення цитокінів (TNF, ІЛ-6, 10 та 1β, TLR4, ендотеліального фактору росту судин і CXCL8), вираженим зниженням експресії eNOS і модуляцією експресії інгібітора активації плазміногена 1 типу (PAI-1). Ці внутрішньоклітинні зміни супроводжуються значним збільшенням проникливості ендотеліального бар’єра, активацією мікротромбоутворення й тяжким перебігом запального процесу [46,47].
Наслідки ендотеліїту при ІХС
Системне пошкодження ендотелію, ендотеліїт є причиною його ремоделювання – стійкої зміни структури та функції, що призводить до втрати контролювальних впливів ЕК із формуванням вираженого зниження судинного тонусу, дифузної зміни перфузії тканин, генералізованого підвищення проникності та гіперкоагуляції [48].
Здатність ендотелію регулювати судинний гомеостаз значною мірою залежить від продукції NO, який синтезується з L-аргініну під дією NOS і низки коферментів. Окислені ЛПНЩ підвищують експресію аргінази та, відповідно, знижують біодоступність аргініну для NOS, сприяючи зменшенню продукції NO, що є основним фактором, який лежить в основі дисфункції ендотелію [49]. На тлі зниження рівня NO в тканинах серця підвищується тонус коронарних судин і знижується вінцевий кровоток. Окрім регуляції судинного тонусу, NO виконує низку інших функцій, а саме знижує адгезію й агрегацію тромбоцитів, адгезію лейкоцитів, запобігає проліферації гладких м’язів, гальмує процеси ремоделювання судинної стінки, знижує виділення прозапальних цитокінів, інгібує експресію молекули клітинної адгезії ICAM-1 (молекули міжклітинної адгезії 1 типу), VCAM-1 (молекули адгезії судинного ендотелію 1 типу) та тканинного фактора, інгібує виділення хемокінів, як-от MCP-1, пригнічує утворення комплексів окислених ЛПНЩ, запобігає вазоконстрикторним ефектам тромбоксану А2, серотоніну, що виділяються з тромбоцитів, знижує стабільність мРНК, моноцитарного хемотаксичного фактора, пригнічує експресію прозапальних генів (NF-κB) та має ангіопротекторні властивості, що робить недостатність NO основною ознакою ендотеліальної дисфункції [50, 51]. Таким чином, формується ланцюжок: ендотеліїт – ендотеліальна дисфункція – дефіцит NO – запуск розвитку атеросклерозу – ішемія та гіпоксія кардіоміоцитів
Дисфункція ендотелію з дефіцитом NO, підвищенням експресії факторів росту, локальних вазоактивних речовин, протеїнів і протеїназ матриксу призводить до судинного ремоделювання, пошкодження структури судини, адгезії моноцитів, що є причиною прогресування атеросклерозу [4].
Наслідком дисфункції ендотелію з порушенням ендотелійзалежного вінцевого кровотоку є розвиток гіпоксії й ішемії міокарда [52]. Крім цього, атеросклероз коронарних артерій безпосередньо впливає на баланс і потребу постачання міокарда киснем, а також є фактором, що становить 90% епізодів ішемії міокарда [53].
Як відомо, оптимальним джерелом енергії для кардіоміоцитів є β-окислення жирних кислот. В умовах гіпоксії й ішемії порушується постачання кисню та поживних речовин до кардіоміоцитів, а тому вони змушені переключатися на анаеробний гліколіз, який дає їм змогу вижити. При цьому в процесі окиснення 1 молекули глюкози утворюється 38 молекул АТФ, тоді як у разі β-окислення найпоширеніших жирних кислот – стеаринової та пальмітинової – може утворитися по 138 молекул АТФ. Отже, серце при ІХС – мотор без палива. Саме тому патогенетично обґрунтованим є застосування препаратів, які впливають на енергетичний обмін міокарда та відновлюють його нормальний окисний шлях синтезу АТФ[54].
Лікування ендотеліїту при ІХС
Зменшення проявів ендотеліїту при ІХС
Потенційно ефективним підходом до корекції дисфункції ендотелію, що виникає на тлі ендотеліїту, є відновлення нормального рівня синтезу NO, зокрема шляхом надання додаткових субстратів для NOS. Таким субстратом є L-аргінін – умовно незамінна амінокислота, котра, будучи попередником NO, відіграє важливу роль у регуляції судинного тонусу та серцево-судинного гомеостазу загалом [51, 55]. Підвищення біодоступності NO не лише забезпечує ендотелійзалежну вазодилатацію, а й зменшує активацію прозапальних генів і експресію молекул ендотеліальної адгезії, впливаючи таким чином на атерогенез [55].
Ефективність застосування L-аргініну в дозі 2 г 3 р/добу протягом 4 тиж для лікування пацієнтів зі стабільною стенокардією I-II функціональних класів (ФК) було доведено в дослідженні A. Palloshi та співавт., у якому продемонстрували його здатність сприяти зниженню ФК стенокардії, систолічного артеріального тиску (АТ) у спокої, поліпшенню якості життя, підвищенню концентрації L-аргініну та циклічного гуанозинмонофосфату [56].
У проспективному подвійному сліпому рандомізованому перехресному дослідженні, проведеному M.R. Adams і співавт., пероральний прийом L-аргініну в дозі 21 г/добу протягом 3 днів сприяв значному покращенню ендотелійзалежної вазодилатації та зниженню адгезії моноцитів до ендотелію (з 50±1 до 42±2%; p<0,01) у хворих з ангіографічно підтвердженим коронарним атеросклерозом [57].
Надалі в рандомізованому подвійному сліпому плацебо-контрольованому перехресному дослідженні з двома періодами лікування (аргінін/плацебо) тривалістю 2 тиж кожний було продемонстровано, що призначення аргініну сприяє покращенню ендотелійзалежної вазодилатації плечових артерій, толерантності до фізичного навантаження та якості життя пацієнтів зі стабільною стенокардією напруження II-III ФК. Зокрема, максимальна вазодилатація плечових артерій у групі аргініну значно зросла порівняно з вихідним рівнем (8,0±4,9 проти 5,5±4,5 відповідно; p=0,004), тоді як у групі плацебо динаміки тонусу судин не було (5,8±5,3 проти 5,8±6,4 до та після призначення плацебо відповідно; p=0,95). Час виконання навантаження збільшився на 16% у період лікування порівняно зі зменшенням на 4% під час періоду плацебо (р=0,05). Крім цього, в групі аргініну спостерігали вірогідне покращення якості життя, оцінену згідно з опитувальником SF-36 (68±13 балів у групі лікування проти 63±21 балів у групі плацебо; р=0,04) [58]. Результати подвійного сліпого плацебо-контрольованого дослідження, що включало 22 пацієнтів зі стабільною стенокардією, свідчать про покращення толерантності до фізичного навантаження лише за 3 дні прийому аргініну [59].
L-аргінін довів свою ефективність у разі його включення в комплексну терапію ішемії міокарда. Зокрема, введення L-аргініну в дозі 3 г/добу протягом 15 днів забезпечувало підвищення активності ферменту супероксиддисмутази, що поглинає вільні радикали, та збільшення рівнів загальних тіолів і аскорбінової кислоти із супутнім зниженням перекисного окиснення ліпідів, умісту карбонілів, сироваткового холестерину й активності прооксидантного ферменту ксантиноксидази [60]. У дослідженні W.H. Yin і співавт. у 31 пацієнта зі стабільною ІХС пероральний прийом L-аргініну в дозі 10 г/добу протягом 4 тиж сприяв поліпшенню ендотеліальної функції та зниженню окислення ЛПНЩ [61].
Метаболізм NO в організмі є циклічним: NO може утворюватися не лише шляхом синтезу з аргініну, але й шляхом його відновлення з нітрит-іону – продукту автоокислення NO й основного резервуару NO в циркуляції. У цьому аспекті ще одним важливим ефектом аргініну є вплив на функцію нирок, а саме на виведення нітритів із сечею. У плацебо-контрольованому дослідженні, проведеному J.Y. Schneider і співавт., пероральне введення аргініну в дозі 10 г/добу впродовж 3-6 міс запобігало втраті біоактивності NO шляхом збільшення реабсорбції нітриту в ниркових канальцях [62].
Нещодавно проведений систематичний огляд підтвердив позитивний ефект добавок аргініну в дозі 3 г/добу при його застосуванні в пацієнтів із серцево-судинними розладами, особливо для запобігання прогресуванню артеріальної гіпертензії й атеросклерозу. Аргінін у дозі 3 г/добу сприяв синтезу NO без розвитку токсичних ефектів [51].
Усунення наслідків ендотеліїту при ІХС – гіпоксії міокарда
L-карнітин (3-гідрокси-4-N,N,N-триметиламінобутират) – це вітаміноподібна модифікована амінокислота, яка відіграє важливу роль у підтримці енергетичного забезпечення організму. Роль L-карнітину в енергетичному обміні міокарда полягає в здатності сприяти перенесенню вільних жирних кислот усередину мітохондрій, забезпечуючи таким чином окисний метаболізм у кардіоміоциті та сприяючи нормальному функціонуванню лівого шлуночка [63,64]. Крім цього, L-карнітин стимулює видалення з мітохондрій продуктів окиснення жирів та інших недоокислених речовин, що відбувається під час ішемічних подій і може призвести до фатальних шлуночкових аритмій [65].
Згідно з результатами метааналізу, який включав 13 плацебо-контрольованих досліджень, застосування L-карнітину знижувало ризик смерті від усіх причин на 27%, шлуночкових аритмій – на 65% і стенокардії – на 40% [66].
Результати ще одного метааналізу 17 рандомізованих контрольованих досліджень, проведеного X. Song і співавт. (2017), підтвердили безпеку й ефективність застосування L-карнітину в пацієнтів із хронічною серцевою недостатністю. L-карнітин сприяв значному підвищенню загальної ефективності, фракції викиду лівого шлуночка, ударного об’єму (8,21 мл), серцевого викиду (0,88 л/хв), а також зменшенню розмірів лівого шлуночка та зниженню рівнів BNP (-124,60 пг/мл) і NT-proBNP (-510,36 пг/мл) у сироватці крові [63].
Крім того, L-карнітин сприяє зниженню рівнів загального холестерину, ЛПНЩ та тригліцеридів, а також підвищенню вмісту ліпопротеїнів високої щільності, що було продемонстровано в метааналізі 55 рандомізованих контрольованих досліджень [67].
L-карнітин має антиоксидантні та протизапальні властивості. Він є прямим антиоксидантом і видаляє вже утворені радикали кисню та пригнічує генерацію радикалів кисню цитозольними й мембранопов’язаними ферментами внаслідок утворення комплексів з іонами заліза та міді в їхніх активних центрах, а також впливає на функціональну активність прозапальних клітин [68]. L-карнітин знижує експресію білка індуцибельної NOS (iNOS) – прозапального ферменту, що генерує велику кількість радикалів NO, які чинять цитотоксичну дію на чужорідні та власні клітини людини; гальмує активність NF-κB, знижує експресію прозапальних цитокінів TNF й ІЛ-6 і збільшує експресію захисного білка PPARα, а також блокує розвиток апоптозу за цим механізмом [68,69].
Тіворель® – ендотеліопротектор і антигіпоксант в одному флаконі, застосування котрого є патогенетично обґрунтованим для лікування пацієнтів з ІХС. У разі хронічної ІХС рекомендованою є така схема застосування розчину Тіворель®: 1 флакон внутрішньовенно краплинно 1 р/добу 10 днів із подальшим переходом на пероральний Тівортін аспартат по 2 мірні ложки 2 р/добу до 2 міс.
Завдяки комплексному складу (2,0 г левокарнітину та 4,2 г L-аргініну гідрохлориду) Тіворель® поліпшує енергозабезпечення серцевого м’яза, посилює окислення вуглеводів у циклі трикарбонових кислот, забезпечує антиоксидантну, антигіпоксичну дії, зменшує прояви ендотеліїту та покращує ендотеліальну функцію, запобігає розвитку та прогресуванню атеросклеротичних бляшок.
Україна належить до країн із дуже високим ризиком розвитку серцево-судинної патології, наслідком чого є зниження працездатності та зростання рівня інвалідності в осіб працездатного віку. У цьому аспекті важливе значення надається своєчасному виявленню категорії осіб із високим кардіоваскулярним ризиком і проведенню належних профілактичних заходів у цій когорті пацієнтів. Інструментом, який допомагає клініцистам оцінити ризик розвитку серцево-судинної події (фатальної чи нефатальної) в найближчі 10 років в умовно здорових осіб (відсутність серцево-судинної патології, стентування/шунтування коронарних судин, цукрового діабету, високого рівня загального холестерину або його фракцій, сімейної гіперхолестеринемії, дуже високого рівня АТ (систолічний АТ – до 179 мм рт. ст.), хронічної хвороби нирок) віком 40-69 років, які мають чинники ризику, є шкала SCORE2 (Systematic COronary Risk Estimation-2). Оцінка серцево-судинного ризику за шкалою SCORE2 проводиться за 5 параметрами, а саме стать, вік, статус куріння, рівень АТ і загального холестерину в ммоль/л. Якщо стать і вік є тими чинниками, які не піддаються модифікації, то на інші три – куріння, рівень АТ і загального холестерину – впливати необхідно. Зокрема, всім пацієнтам рекомендуються відмова від тютюнокуріння, корекція підвищеного АТ і дисліпідемії [70]. Також варто зазначити, що ці три чинники ризику перегукуються в контексті розвитку ендотеліальної дисфункції. Саме тому доцільним є призначення розчину Тіворель® для корекції проявів ендотеліальної дисфункції та профілактики розвитку кардіоваскулярних подій пацієнтам високого ризику за шкалою SCORE2.
Автори: Тащук В.К., д.м.н., професор, завідувач кафедри внутрішньої медицини, фізичної реабілітації, спортивної медицини Буковинського державного медичного університету, м. Чернівці.
Література:
- Уніфікований клінічний протокол первинної, вторинної (спеціалізованої) та третинної (високоспеціалізованої) медичної допомоги (УКПМД) «Стабільна ішемічна хвороба серця»,2021
- Libby, P., Buring, J. E., Badimon, L., Hansson, G. K., Deanfield, J., Bittencourt, M. S., … Lewis, E. F. (2019). Atherosclerosis. Nature Reviews Disease Primers, 5(1). doi:10.1038/s41572-019-0106-z 10.1038/s41572-019-0106-z
- Ammirati, E.; Moroni, F.; Magnoni, M.; Camici, P.G. The role of T and B cells in human atherosclerosis and atherothrombosis. Clin. Exp. Immunol. 2015, 179, 173–187.
- В.О. Шумаков, Атеросклероз як клінічний прояв ендотеліальної дисфункції. Медична газета «Здоров’я України 21 сторіччя», тематичний номер «Кардіологія, Ревматологія, Кардіохірургія» 2020
- Gary H. Gibbons,Endothelial function as a determinant of vascular function and structure: A new therapeutic target, The American Journal of Cardiology, Volume 79, Issue 5, Supplement 1, 1997,Pages 3-8, ISSN 0002-9149, org/10.1016/S0002-9149(97)00122-7. № 1 (68), 2020 р
- О. А. Воробьева Терапевтические эффекты метаболитотропного кардиопротектора, содержащего L-аргинин и инозин, у больных пожилого и старческого возраста с ИБС. Патологія. 2012. 2. 98-101.
- Бабак, О.Я., Молодан, В.І. Просоленко, К.О.Железнякова, Н.М.Андреева, А.О.Зелена, І.І. Оптимізація діагностики функціонального стану ендотелію у хворих на гіпертонічну хворобу у поєднанні з ожирінням, методичні рекомендації 2014. repo.knmu.edu.ua/handle/123456789/9992
- Кравчун, П., Шелест, М., Ковальова, Ю., Шелест, Б., & Риндіна, Н. (2013). Особливості змін маркерів запалення у хворих на ішемічну хворобу серця з ожирінням. Медицина сьогодні і завтра, 59(2), 38-42. msz.knmu.edu.ua/article/view/145.
- В.І. Денисюк, О.В. Ковальчук, О.В. Денисюк. Ендотеліальна функція судин при ішемічній хворобі серця у поєднанні з артеріальною гіпертензією Практична ангіологія. 2008 6(17).
- Severino, P.; D’Amato, A.; Pucci, M.; Infusino, F.; Adamo, F.; Birtolo, L.I.; Netti, L.; Montefusco, G.; Chimenti, C.; Lavalle, C.; Maestrini, V.; Mancone, M.; Chilian, W.M.; Fedele, F. Ischemic Heart Disease Pathophysiology Paradigms Overview: From Plaque Activation to Microvascular Dysfunction. Int. J. Mol. Sci. 2020, 21, 8118. doi.org/10.3390/ijms21218118
- Аксьонов Є. В. Ендотеліальна дисфункція та шляхи її профілактики при проведенні рентгенендоваскулярних процедур по реканалізації коронарних артерій. Український журнал медицини, біології та спорту – Том 4, № 5 (21) doi: 10.26693/jmbs04.05.102
- Wang K, Zuo G, Zheng L, Zhang C, Wang D, Cao Z, et al. Effects of tirofiban on platelet activation and endothelial function in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention. Cell Biochem Biophys. 2015; 71(1): 135-42. PMID: 25123839. DOI: 10.1007/s12013-014-0173-4
- Steinberg, D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as partners in crime. Nat. Med. 2002, 8, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kohno, H.; Hasegawa, A.; Toshima, S.; Amaki, T.; Kurabayashi, M.; Nagai, R.; Suzuki, T.; Amaki, T.; Nagai, R.; et al. Diagnostic implications of circulating oxidized low density lipoprotein levels as a biochemical risk marker of coronary artery disease. Clin. Biochem. 2002, 35, 347–353. [Google Scholar] [CrossRef]
- Mehta, J.L.; Li, D. Identification, regulation and function of a novel lectin-like oxidized low-density lipoprotein receptor. J. Am. Coll. Cardiol. 2002, 39, 1429–1435. [Google Scholar] [CrossRef][Green Version]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Prinzmetal, M.; Kennarner, R.; Merliss, B.; Wads, T.; Bor, N. Angina pectoris. I. A variant form of angina pectoris; preliminary report. Am. J. Med. 1959, 27, 375–388. [Google Scholar] [CrossRef]
- Maseri, A. Mechanisms of myocardial ischemia. Cardiovasc. Drugs Ther. 1990, 4 (Suppl. 4), 827–831. [Google Scholar] [CrossRef]
- Wu, N.; Li, W.; Shu, W.; Lv, Y.; Jia, D. Inhibition of Rho-kinase by fasudil restores the cardioprotection of ischemic postconditioninng in hypercholesterolemic rat heart. Mol. Med. Rep. 2014, 10, 2517–2524. [Google Scholar] [CrossRef][Green Version]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Grillo, R.L.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N. Engl. J. Med. 1994, 331, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Ehara, S.; Ueda, M.; Naruko, T.; Haze, K.; Itoh, A.; Otsuka, M.; Komatsu, R.; Matsuo, T.; Itabe, H.; Takano, T.; et al. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation 2001, 103, 1955–1960. [Google Scholar] [CrossRef]
- Mollace, V.; Gliozzi, M.; Musolino, V.; Carresi, C.; Muscoli, S.; Mollace, R.; Tavernese, A.; Gratteri, S.; Palma, E.; Morabito, C.; et al. Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: Role of oxidative stress and LOX-1 receptor expression. Int. J. Cardiol. 2015, 184, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Medina J, Muñoz M, Rodriguez C, Sanchez A, Contreras C, Carballido-Rodríguez J, Prieto D. Endothelial Dysfunction: An Intermediate Clinical Feature between Urolithiasis and Cardiovascular Diseases. Int J Mol Sci. 2022 Jan 14;23(2):912. doi: 10.3390/ijms23020912. PMID: 35055099; PMCID: PMC8778796
- Hattori Y, Hattori K, Machida T, Matsuda N. Vascular endotheliitis associated with infections: Its pathogenetic role and therapeutic implication. Biochem Pharmacol. 2022 Mar;197:114909. doi: 10.1016/j.bcp.2022.114909. Epub 2022 Jan 10. PMID: 35021044; PMCID: PMC8743392.
- Busse, I. Fleming, Vascular endothelium and blood flow, Handb. Exp. Pharmacol. 176 (pt 2) (2006) 43–78.
- Godo, H. Shimokawa, Endothelial functions, Arterioscler. Thromb. Vasc. Biol. 37 (9) (2017) e108–e114.
- Krüger-Genge, A. Blocki, R.-P. Franke, F. Jung, Vascular endothelial cell biology: an update, Int. J. Mol. Sci. 20 (2019) 4411.
- Davey MP, Martin TM, Planck SR, Lee J, Zamora D, Rosenbaum JT: Human endothelial cells express NOD2/CARD15 and increase IL-6 secretion in response to muramyl dipeptide. Microvasc Res 2006, 71(2):103–107
- Krausgruber T, Fortelny N, Fife-Gernedl V et al.Structural cells are key regulators of organ-specific immune responses // Nature. 2020;583(7815):296–302. Doi: 10.1038/s41586-020-2424-4.
- Armingol E, Officer A, Harismendy O, Lewis NE.Deciphering cell-cell interactions and communication from gene expression // Nat. Rev. Genet. 2021;22(2):71–88. Doi: 10.1038/s41576-020-00292-x.
- Kircheis R, Haasbach E, Lueftenegger D, Heyken WT, Ocker M, Planz O. NF-κB Pathway as a Potential Target for Treatment of Critical Stage COVID-19 Patients. Front Immunol. 2020 Dec 10;11:598444. doi: 10.3389/fimmu.2020.598444. PMID: 33362782; PMCID: PMC7759159.
- Приходько-Дибська К. COVID‑19: результати аутопсії [Електронний ресурс] / Режим доступу: www.umj.com.ua/article/183365/covid‑19-rezultati-autopsiyi
- Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020 May 2;395(10234):1417-1418. doi: 10.1016/S0140-6736(20)30937-5. Epub 2020 Apr 21. PMID: 32325026; PMCID: PMC7172722.
- Suzuki T, Ohashi Y. Corneal endotheliitis. Semin Ophthalmol. 2008 Jul-Aug;23(4):235-40. doi: 10.1080/08820530802111010. PMID: 18584561.
- Chudek J. Adipose tissue, inflammation and endothelial dysfunction / J. Chudek, A. Wiecek // Pharmacological Reports. – 2006. – Vol. 58, Suppl. – P. 81-88
- Versari D. Endothelial dysfunction as a target for preventation of cardiovascular disease / D. Versari, E. Daghini, A. Virdis // Diabetes Care. – 2009. – Vol. 32. – P. 314-321.
- Т.О. Проскура.Потенційні органи-мішені постковідного синдрому. ЖУРНАЛ НЕВРОЛОГІЇ ім. Б.М. Маньковського’ 2021, ТОМ 9, № 1-2 neuroscience.com.ua/index.php/journal/article/download/372/303/
- Becker RC. COVID-19-associated vasculitis and vasculopathy // J. Thromb. Thrombolysis. 2020;50(3):499–511.
- McGonagle D, Bridgewood C, Ramanan AV, Meaney JFM, Watad A. COVID-19 vasculitis and novel vasculitis mimics // Lancet Rheumatol. 2021;3(3):e224–e233.
- Seeßle J, Waterboer T, Hippchen T, Simon J, Kirchner M, Lim A, Müller B, Merle U. Persistent Symptoms in Adult Patients 1 Year After Coronavirus Disease 2019 (COVID-19): A Prospective Cohort Study. Clin Infect Dis. 2022 Apr 9;74(7):1191-1198. doi: 10.1093/cid/ciab611. PMID: 34223884; PMCID: PMC8394862.
- Doykov I, Hällqvist J, Gilmour KC, Grandjean L, Mills K, Heywood WE. ‘The long tail of Covid-19’ – The detection of a prolonged inflammatory response after a SARS-CoV-2 infection in asymptomatic and mildly affected patients. F1000Res. 2020 Nov 19;9:1349. doi: 10.12688/f1000research.27287.2. PMID: 33391730; PMCID: PMC7745182.
- Fogarty H, Karampini E, O’Donnell AS, Ward SE, O’Sullivan JM, O’Donnell JS; Irish COVID-19 Vasculopathy Study (iCVS) investigators. Persistent endotheliopathy in the pathogenesis of long COVID syndrome – Reply to comment from von Meijenfeldt et al. J Thromb Haemost. 2022 Jan;20(1):270-271. doi: 10.1111/jth.15578. Epub 2021 Nov 22. PMID: 34738307; PMCID: PMC8646468.
- Loktionova IL, Pokrovskiy MV, Ragulina VA, Titareva LV, Denisuk ТА, Stupakova EV,Sytnik МV, Saroyan KV, Losenok SА. The status of vascular endotheliumfunction in infectious diseases of various etiologies // Nauchnye vedomosti, Seriya Medicina. Farmaciya. 2012. No 4 (123);17/1: 20–31
- Kühnl A, Rien C, Spengler K, Kryeziu N, Sauerbrei A,Heller R, Henke A. Characterization of coxsackievirus B3replication in human umbilical vein endothelial cells // Med Microbiol Immunol. 2014;203(4):217–229. Doi: 10.1007/s00430-014-0333-6. PMID: 24615265.
- Han T, Lai Y, Jiang Y, Liu X, Li D. Influenza A virus infects pulmonary microvascular endothelial cells leading tomicrovascular leakage and release of pro-inflammatory cytokines // Peer J. 2021;(9):e11892. Doi: 10.7717/peerj.11892.PMID: 34414033; PMCID: PMC8344683.
- Han T, Lai Y, Jiang Y, Liu X, Li D. Influenza A virus infects pulmonary microvascular endothelial cells leading to microvascular leakage and release of pro-inflammatory cytokines // Peer J. 2021;(9):e11892. Doi: 10.7717/peerj.11892.PMID: 34414033; PMCID: PMC8344683.
- Herold S, Becker C, Ridge KM, Budinger GR. Influenza virus-induced lung injury: pathogenesis and implications for treatment // Eur Respir J. 2015;45(5):1463–1478. Doi:10.1183/09031936.00186214. PMID: 25792631.
- De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005 Oct;25(10):2106-13. doi: 10.1161/01.ATV.0000181743.28028.57. Epub 2005 Aug 11. PMID: 16100037.
- Ryoo, S., Berkowitz, D. E., & Lim, H. K. (2011). Endothelial arginase II and atherosclerosis. Korean Journal of Anesthesiology, 61(1), 3-11.
- В. Курята, Ю. С. Кушнір, Т. О. Віхрова. Медицина невідкладних станів. 2019. 3. 45-50. nbuv.gov.ua/UJRN/Medns_2019_3_9.
- Gambardella J, Khondkar W, Morelli MB, Wang X, Santulli G, Trimarco V. Arginine and Endothelial Function. Biomedicines. 2020; 8(8):277. doi.org/10.3390/biomedicines8080277
- Hasdai D, Gibbons RJ, Holmes DR Jr, Higano ST, Lerman A. Coronary endothelial dysfunction in humans is associated with myocardial perfusion defects. Circulation. 1997 Nov 18;96(10):3390-5. doi: 10.1161/01.cir.96.10.3390. PMID: 9396432
- Orsini E, Zito GB. Matching pathophysiology and evidence-based medicine for optimal management of ischemic heart disease. J Cardiovasc Med (Hagerstown). 2010;11(6):469-479
- В.О. Шумаков, Можливі шляхи ефективної енергетичної підтримки міокарда. Медична газета «Здоров’я України 21 сторіччя», № 5-6. 2022
- Luiking YC, Ten Have GA, Wolfe RR, Deutz NE. Аргінін de novo та утворення оксиду азоту в хворобливих станах. Am. J. Physiol. ендокринол. Метаб. 2012 рік; 303 :E1177–E1189. doi: 10.1152/ajpendo.00284.2012
- Palloshi A, Fragasso G, Piatti P, Monti LD, Setola E, Valsecchi G, Galluccio E, Chierchia SL, Margonato A. Effect of oral L-arginine on blood pressure and symptoms and endothelial function in patients with systemic hypertension, positive exercise tests, and normal coronary arteries. Am J Cardiol. 2004 Apr 1;93(7):933-5. doi: 10.1016/j.amjcard.2003.12.040. PMID: 15050504.
- Adams MR, McCredie R, Jessup W, Robinson J, Sullivan D, Celermajer DS. Oral L-arginine improves endothelium-dependent dilatation and reduces monocyte adhesion to endothelial cells in young men with coronary artery disease. Atherosclerosis. 1997 Mar 21;129(2):261-9. doi: 10.1016/s0021-9150(96)06044-3. PMID: 9105569.
- Maxwell AJ, Zapien MP, Pearce GL, MacCallum G, Stone PH. Randomized trial of a medical food for the dietary management of chronic, stable angina. J Am Coll Cardiol. 2002 Jan 2;39(1):37-45. doi: 10.1016/s0735-1097(01)01708-9. PMID: 11755284.
- Ceremuzynski L., Chamiec T., Herbaczynska-Cedro K. Effect of supplemental oral L-arginine on exercise capacity in patients with stable angina pectoris. Am. J. Cardiol. 1997;80:331–333. doi: 10.1016/S0002-9149(97)00354-8. [PubMed] [CrossRef] [Google Scholar] [Ref list]
- Tripathi P, Misra MK. Therapeutic role of L-arginine on free radical scavenging system in ischemic heart diseases. Indian J Biochem Biophys. 2009 Dec;46(6):498-502. PMID: 20361713.
- Yin, W. H., Chen, J. W., Tsai, C., Chiang, M. C., Young, M. S., & Lin, S. J. (2005). L-arginine improves endothelial function and reduces LDL oxidation in patients with stable coronary artery disease. Clinical Nutrition, 24(6), 988-997.
- Schneider, J.Y., Rothmann, S., Schröder, F. et al. Effects of chronic oral L-arginine administration on the L-arginine/NO pathway in patients with peripheral arterial occlusive disease or coronary artery disease: L-Arginine prevents renal loss of nitrite, the major NO reservoir. Amino Acids 47, 1961–1974 (2015). org/10.1007/s00726-015-2031-0
- Song X, Qu H, Yang Z, Rong J, Cai W, Zhou H. Efficacy and Safety of L-Carnitine Treatment for Chronic Heart Failure: A Meta-Analysis of Randomized Controlled Trials. Biomed Res Int. 2017;2017:6274854. doi: 10.1155/2017/6274854. Epub 2017 Apr 13. PMID: 28497060; PMCID: PMC5406747.
- В.О, Шумаков- Ефективність терапії пацієнтів із ішемічною хворобою серця з застосуванням фіксованої комбінації L-аргініну та L-карнітину з точки зору доказової медицини. Огляд міжнародних наукових джерел. Ukrainian Medical Journal.2021.143. 10.32471/umj.1680-3051.143.207515
- В.К. Тащук Медична газета «Здоров’я України 21 сторіччя», тематичний номер «Кардіологія, Ревматологія, Кардіохірургія» 2020
- DiNicolantonio JJ, Lavie CJ, Fares H, Menezes AR, O’Keefe JH. L-carnitine in the secondary prevention of cardiovascular disease: systematic review and meta-analysis. Mayo Clin Proc. 2013 Jun;88(6):544-51. doi: 10.1016/j.mayocp.2013.02.007. Epub 2013 Apr 15. PMID: 23597877.
- Askarpour M, Hadi A, Miraghajani M, Symonds ME, Sheikhi A, Ghaedi E. Beneficial effects of l-carnitine supplementation for weight management in overweight and obese adults: An updated systematic review and dose-response meta-analysis of randomized controlled trials. Pharmacol Res. 2020 Jan;151:104554. doi: 10.1016/j.phrs.2019.104554. Epub 2019 Nov 17. PMID: 31743774.
- І.О. Мітюряєва – Корнійко Імуномодулюючі можливості l-карнітіну – іноваційне медикаментозне супроводження терапії інфекційногопроцесу. Міжнародний журнал педіатрії, акушерства та гінекології Липень/Вересень 2018 Том 12 №3
- Koc A, Ozkan T, Karabay AZ, Sunguroglu A, Aktan F. Effect of L-carnitine on the synthesis of nitric oxide in RAW 264·7 murine macrophage cell line. Cell Biochem Funct. 2011 Dec;29(8):679-85. doi: 10.1002/cbf.1807. Epub 2011 Oct 19. PMID: 22012571.
- SCORE2 risk prediction algorithms:new models to estimate 10-year risk of cardiovascular disease in Europe European Heart Journal (2021) 00, 1–16 CLINICAL RESEARCH doi:10.1093/eurheartj/ehab309